





Companies leading in aerospace, energy, and computing seek innovative materials to enhance performance. However, understanding the actual behavior of these materials in practical applications like rockets or computer chips requires creating and testing them. This necessity arises because even advanced simulation methods find it challenging to accurately model complex chemical structures in modern solid materials, increasing costs and time for innovation. MIT researchers have now developed a method to precisely simulate metal behavior, regardless of chemical complexity. This is achieved through machine-learning models that enhance the speed and accuracy of material simulations by using training datasets that reflect diverse atomic environments in chemically disordered materials.
A recent paper in Science Advances demonstrates that this method can accurately predict the properties of diverse metal alloys under various conditions, aiding in the development of new materials, particularly where experimentation is costly. “The focus of the paper is metallic alloys, which is the field I work in, but this could be adapted to other types of materials, like semiconductors,” explains senior author Rodrigo Freitas, MIT’s TDK Career Development Professor in Materials Science and Engineering. Freitas emphasizes the method’s adaptability to create new sustainable materials, such as steels for aerospace applications. The paper includes contributions from first author Killian Sheriff PhD ’26, MIT PhD students Daniel Xiao and Yifan Cao, and University of Sheffield Senior Lecturer Lewis R. Owen.
Material properties are largely determined by the internal arrangement of their chemical elements. Although two materials may have identical chemical compositions, their differing arrangements can lead to one being brittle and the other capable of deformation without breaking. Simulating these differences requires modeling materials atom by atom, using models that explain atomic interactions. Over the past twenty years, machine learning has become the most precise method for creating these models, especially when dealing with unordered atomic arrangements, which are common in solid materials. “The real challenge in our field is modelling these chemically disordered phases,” Freitas notes, highlighting the difficulty for machine-learning models to learn from the vast variety of local chemical environments present in such materials.
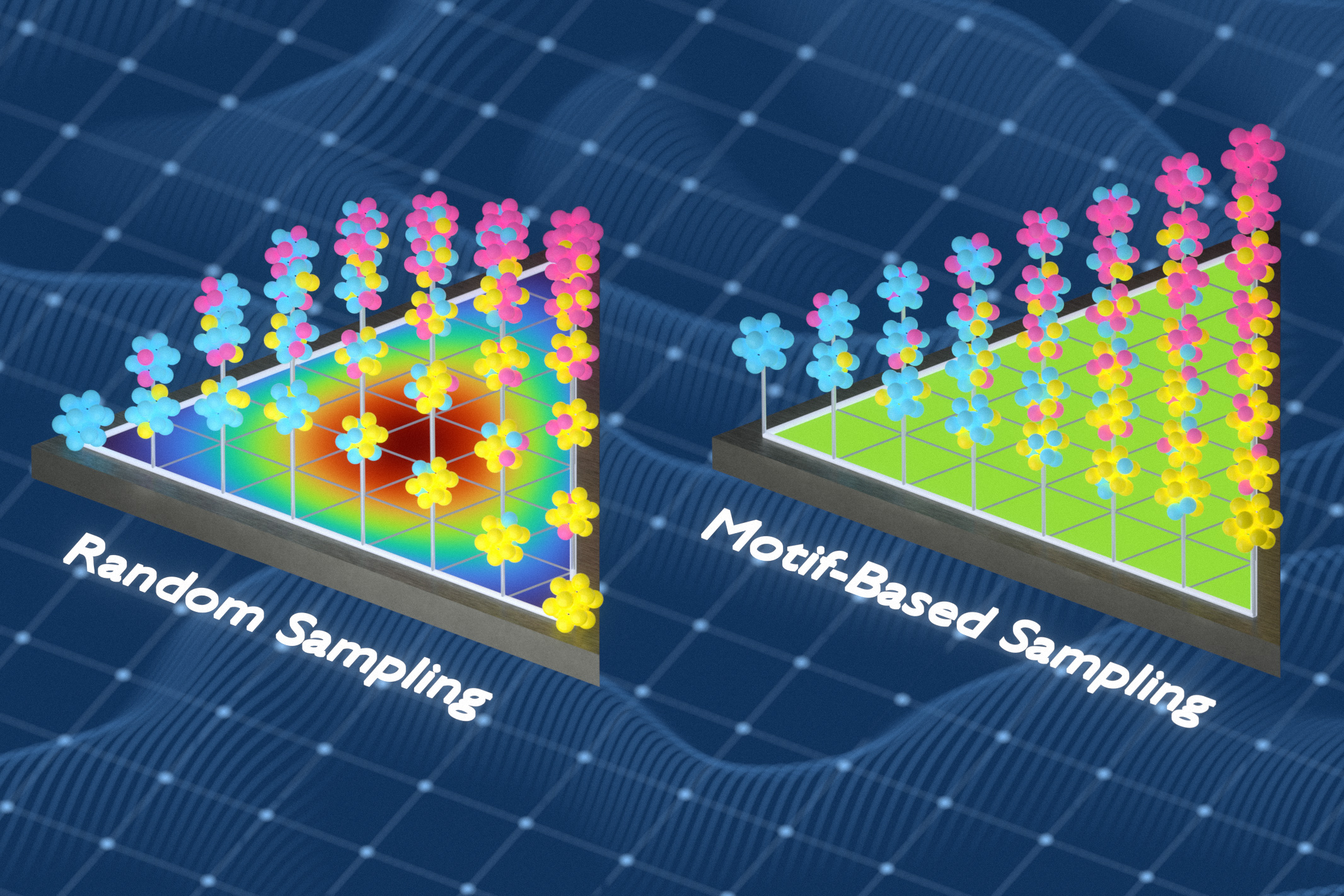
The main issue is the lack of representative training data for atom-by-atom simulations, with current methods often demanding over 100,000 hours of computation to generate data for a single material. These methods also struggle to adapt when the material composition changes. Freitas’ team previously developed a technique to measure the chemical complexity of solid materials by analyzing atomic group frequency and spacing. In this study, they used this technique to create better training datasets, leveraging information theory to generate data that captures a wider range of local chemical environments. The method involves swapping out atoms in samples to minimize repetition and expose models to new chemical environments.
Freitas explains, “We kept optimizing the training set so it captured as many different local environments as possible.” By replacing redundant examples with new ones, the training set becomes more informative, each entry offering fresh information. Models trained with these datasets predicted material properties more accurately than those trained with random sampling or other popular methods. “The starting point for all these atom-by-atom simulations is: Are you able to accurately describe the chemical bond between atoms?” Freitas states, highlighting the importance of accurate chemical descriptions in simulations to reflect real-world material behavior.
The researchers applied their technique to create machine-learning training datasets for a group of chemically diverse metal alloys, demonstrating their models’ superior accuracy compared to larger models from companies like Google and Microsoft. “We got to a point where we were convinced it worked without using these expensive brute-force methods,” Freitas says. He encouraged Sheriff to test the models’ ability to accurately predict useful material properties, which Sheriff did extensively with Xiao and Cao, comparing simulations against real atomic ordering measurements from Owen’s experimental data.
The method effectively captures hidden patterns in sample data, described in the paper as “subtle energetic biases toward certain local chemical configurations.” These small energetic differences are crucial in determining which phases form in an alloy and how they change with temperature and composition, ultimately affecting material properties. Daniel Xiao led simulations that showed the team’s models could predict phase diagrams matching experimental data. Such diagrams are crucial for designing and processing alloys, mapping stable phases across different temperatures and compositions. “Phase diagrams are one of the main ways people connect materials modeling to real processing decisions,” Freitas notes.
The team is now using their approach to study how altering an alloy’s composition influences mechanical properties and radiation tolerance, aiming to design materials that remain robust in harsh environments. They are also working to integrate the method with existing industry tools and workflows. “Industry isn’t going to change the way they do things if what you’re creating doesn’t fit into their existing operating procedures,” Freitas states, emphasizing the need for practical applicability in material design decisions. The research received support from the U.S. Air Force Office of Scientific Research.
Original Source: news.mit.edu



